Sans gêne
04/08/2020On parle beaucoup ces derniers temps des kits de test ADN récréatifs, dont l'utilisation est encore interdite en France. Leur diffusion est néanmoins peu entravée, des entreprises étrangères spécialisées adressant discrètement des colis à quiconque peut les dénicher sur le web et souhaite sonder ses allèles. La prudence est de mise, le privilège n'étant pas accordé à titre grâcieux par ces entités, qui se réservent d'ailleurs le droit de partager tout ou partie des génotypes obtenus avec d'autres curieux bien nantis. La situation pourrait évoluer avec les débats à venir sur la loi de bioéthique. Voilà qui conclut notre épisode de Black Mirror pour ce soir, merci.
Les enseignements tirés de recherches scientifiques en la matière, encadrées et à plus grand échelle, restent extraordinaires. Les deux exemples que j'évoque ci-dessous sont tout à fait réjouissants, avec une portée anthropologique et historique.
Répartition géniale de populations diverses (RGPD)
Il y a une dizaine d'années, une équipe de chercheurs menée par John Novembre s'est penchée sur la diversité génétique des populations sur le continent européen. Le génome de 1387 individus a été étudié en détail, et comparé en 200000 positions fixes, dites loci. Chaque locus en question pointe vers un polymorphisme d'un seul nucléotide, ou SNP1: la variation d'une unique paire de bases azotées sur un brin d'ADN, identifiée chez au moins 1% de la population humaine. 90% des variations génétiques humaines s'expliquent par ces polymorphismes. Les progrès récents en génomique ont permis une grande automatisation de telles études, grâce notamment à l'utilisation de puces à ADN.
Une origine ancestrale récente était également définie pour chaque individu: le pays où vécurent ses grands-parents si l'information était disponible2, ou à défaut celui de sa naissance.
L'analyse en composante principale (PCA3) fut mise à contribution pour tirer la substantifique moëlle de ces mesures. On cherche grâce à cette méthode statistique à décrire les données relevées pour chaque individu grâce à de nouvelles variables (les composantes principales), choisies pour être indépendantes les unes des autres et présenter au mieux la diversité dans la population observée. Le côté génial de cette approche est qu'elle permet de résumer l'information, en se concentrant sur les composantes apportant le plus de détails, et en négligeant les autres, jugées redondantes ou bruitées.
Dans le cas présent, l'équipe a obtenu des résultats remarquables en ne retenant que les 2 premières composantes principales, notées PC1 et PC2: la projection des résultats s'apparente à une carte géographique de l'Europe relativement fidèle, moyennant une légère rotation des axes (illustration a ci-dessous).
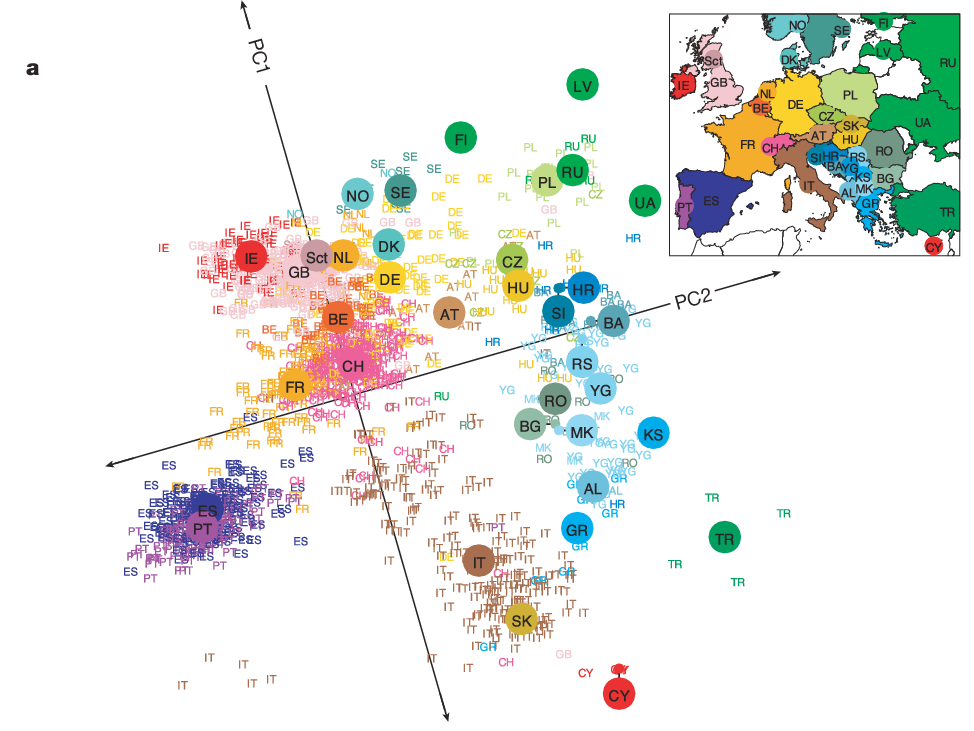
Chaque point correspond à un individu, et son origine ancestrale déclarée est précisée par le code couleur défini dans la carte miniature en haut à droite de l'illustration. Les pastilles sur fond uni représentent les valeurs médianes des composantes PC1 et PC2 par pays ou région.
La distance entre deux points sur ce schéma traduit la similarité génétique des deux personnes qu'elles représentent. Des ensembles de population se dégagent, avec des concentrations de points pour les individus d'un pays donné. On constate des recouvrements entre pays voisins, et deux principaux écarts avec les positions géographiques attendues: pour la Slovaquie en particulier et la Russie dans une moindre mesure. La Slovaquie est représentée par un unique individu dans l'étude, avec un résultat laissant à penser qu'il dispose en fait d'aïeux italiens. La Russie apparaît elle trop à "l'ouest" dans le plan PC1-PC2, peut-être à cause d'un déséquilibre dans le découpage géographique choisi: le pays est representé d'un seul bloc, malgré une surface plus vaste que l'ensemble des autres régions considérées.
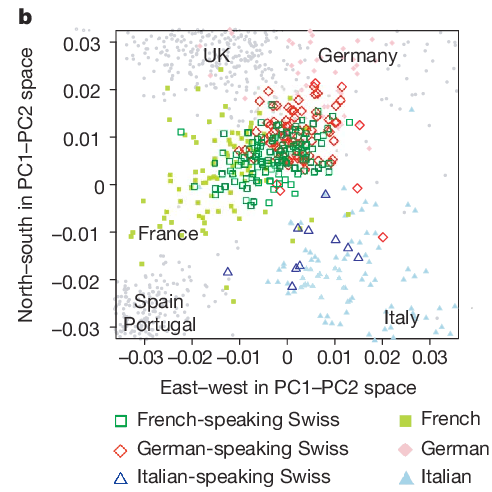
L'illustration b ci-dessus montre un exemple inverse, où des informations démographiques supplémentaires (et un échantillon étendu) permettent d'affiner les conclusions: il est possible de différencier la population suisse assez finement, en fonction des langues maternelles (Français, Allemand ou Italien4) et de rapprocher leurs locuteurs respectifs des populations des pays frontaliers.
Une fois le plan PC1-PC2 établi, il est alors envisageable d'y projeter le génotype d'un individu ne figurant pas dans l'échantillon de départ: ses "coordonnées" sont obtenus en appliquant la même transformation qu'aux données d'entrée. On peut enfin attribuer une origine ancestrale présumée, en assignant le point obtenu à la région génétiquement la plus proche: on parle de classification supervisée. Les kits ADN commerciaux s'appuient sur de telles techniques pour éclairer la généalogie de leurs utilisateurs. Toutefois, l'accès aux modèles expérimentaux et à leurs données d'entrée n'est pas nécessairement garanti par les entreprises proposant ce genre de services, là où le système de peer-review est censé assurer transparence et falsifiabilité pour les articles scientifiques publiés5. Charge aux particuliers de faire confiance aux tests qu'ils entreprennent!
L'Ibère vient
Un point frappant sur la carte présentée plus haut est la dispersion des points rattachés à l'Espagne et au Portugal: une grande diversité génétique caractérise la péninsule ibérique. Ceci a récemment conduit une équipe de chercheurs d'Oxford et de l'Université de Santiago de Compostela à la Corogne à explorer la structure des populations à l'échelle de ces deux pays. L'excellente émission de la BBC Inside Science avait invité Clare Bycroft, qui a dirigé l'étude, à discuter de leurs résultats en février 2019.
Une première étude portait sur 1413 individus, uniquement espagnols, génotypés en un peu moins de 700000 SNP. Les individus ont été regroupés en fonction de la quantité de matériel génétique qu'ils sont susceptibles d'avoir hérité d'un ancêtre commun. Un arbre hiérarchique (ou phylogénétique) a ensuite été construit de proche en proche, en appariant ces groupes de sorte à maximiser la probabilité qu'ils soient eux-mêmes issus d'une population ancestrale commune, puis en réitérant cette étape au niveau supérieur jusqu'à couvrir tout l'échantillon.6 L'illustration 2.a ci-dessous présente cet arbre, au niveau intermédiaire où 14 groupes existent (145 existent au niveau le plus bas de l'arbre, mais ne sont que peu représentatifs individuellement, compte tenu du faible nombre d'invididus qu'ils incluent). La désignation de ces groupes fait référence au lieu d'origine de la majorité de leurs individus, mais leur construction n'a effectivement dépendu que d'informations génétiques, et pas géographiques.
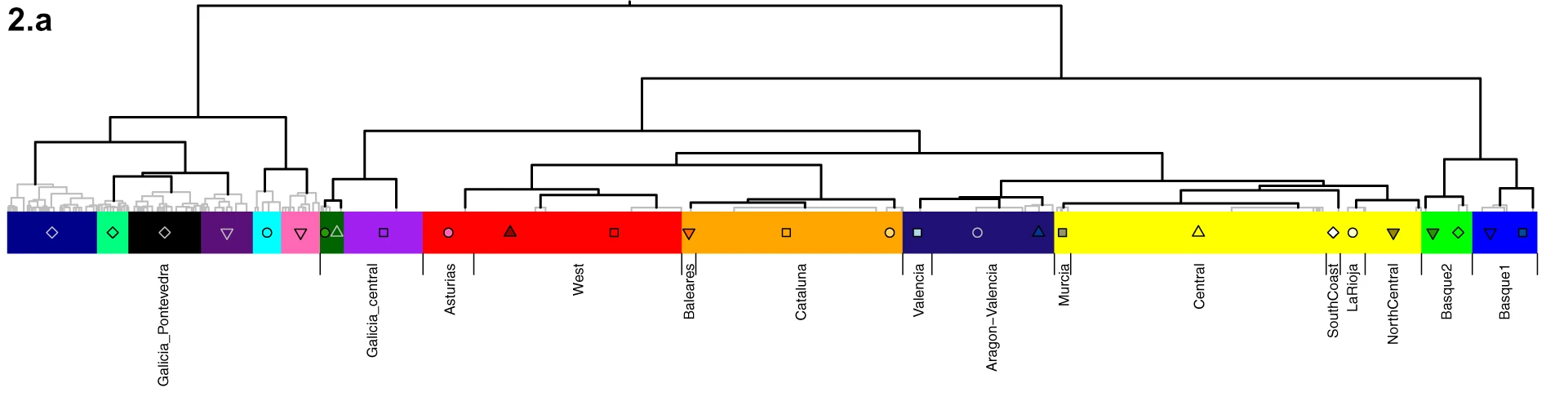
Ces données géographiques furent exploitées dans un second temps, pour établir la carte 2.b: chaque individu est représenté par un point placé à la moyenne des coordonnées des lieux de naissance de ses 4 grand-parents, et sa couleur est celle du groupe auquel il appartient, comme défini dans l'arbre phylogénétique. Enfin, le fond de la carte est coloré en fonction des contributions respectives des différents groupes, en considérant une grille faite de carrés de 3km de côté (de bons gros pixels). Qu'observe-t-on? De nettes bandes verticales, ne suivant pas les contours actuels des communautés autonomes du pays, également representées.
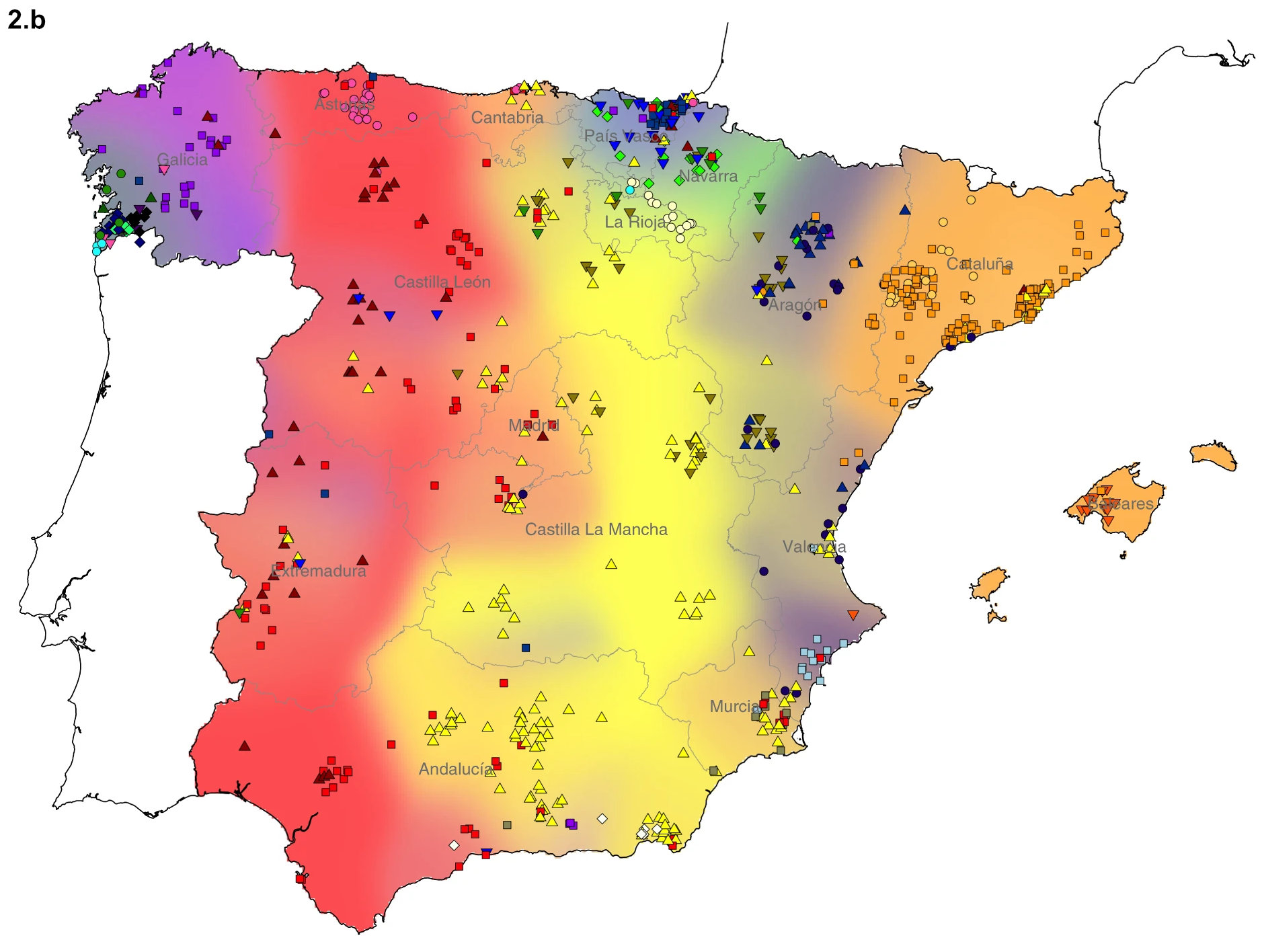
Ces bandes seraient un effet des mouvements de population associés à la conquête musulmane, puis à la Reconquista, entre les VIIIème et XIIIème siècles de notre ère. Les royaumes catholiques, cantonnés au nord de la péninsule au plus fort de l'expansion des Omeyyades, se sont progressivement étendus vers le sud. La solidarité fut toute relative entre ces alliés du moment, ce grand élan ayant aussi été pour eux l'occasion d'ajuster ici et là leurs frontières latérales. Mais le partage du pouvoir politique dans la région au début du XIIIème montre bien un étalement vertical des différentes factions. Cette structure en bandes est encore plus évidente pour la diffusion des langues du nord au sud, marqueur classique de l'avancée de la reconquête (voir l'illustration 2.c). Clare Bycroft et son équipe ont ensuite réussi à établir que la localisation de deux invididus dans une même région linguistique historique est un bien meilleur prédicteur d'appartenance au même groupe génétique que ne le sont la distance physique entre ces individus, ou le fait de vivre dans la même communauté autonome.
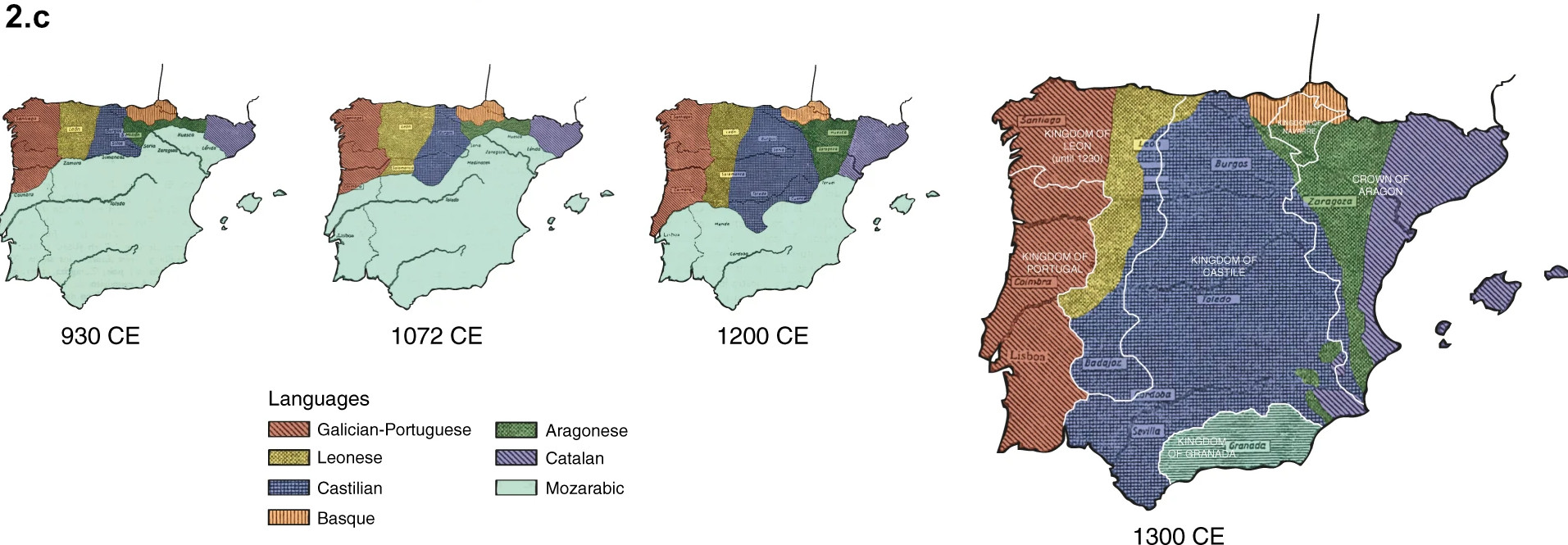
Une seconde analyse incluait cette fois des individus portugais, malheureusement sans informations géographiques aussi précises que pour l'Espagne7. La même structure en bandes verticales s'observe dans ce cas plus large (carte 2.d ci-dessous): les Portugais sont ainsi plus proches génétiquement des Galiciens que de leurs voisins à l'est, Castillans, Estrémègnes et Andalous.
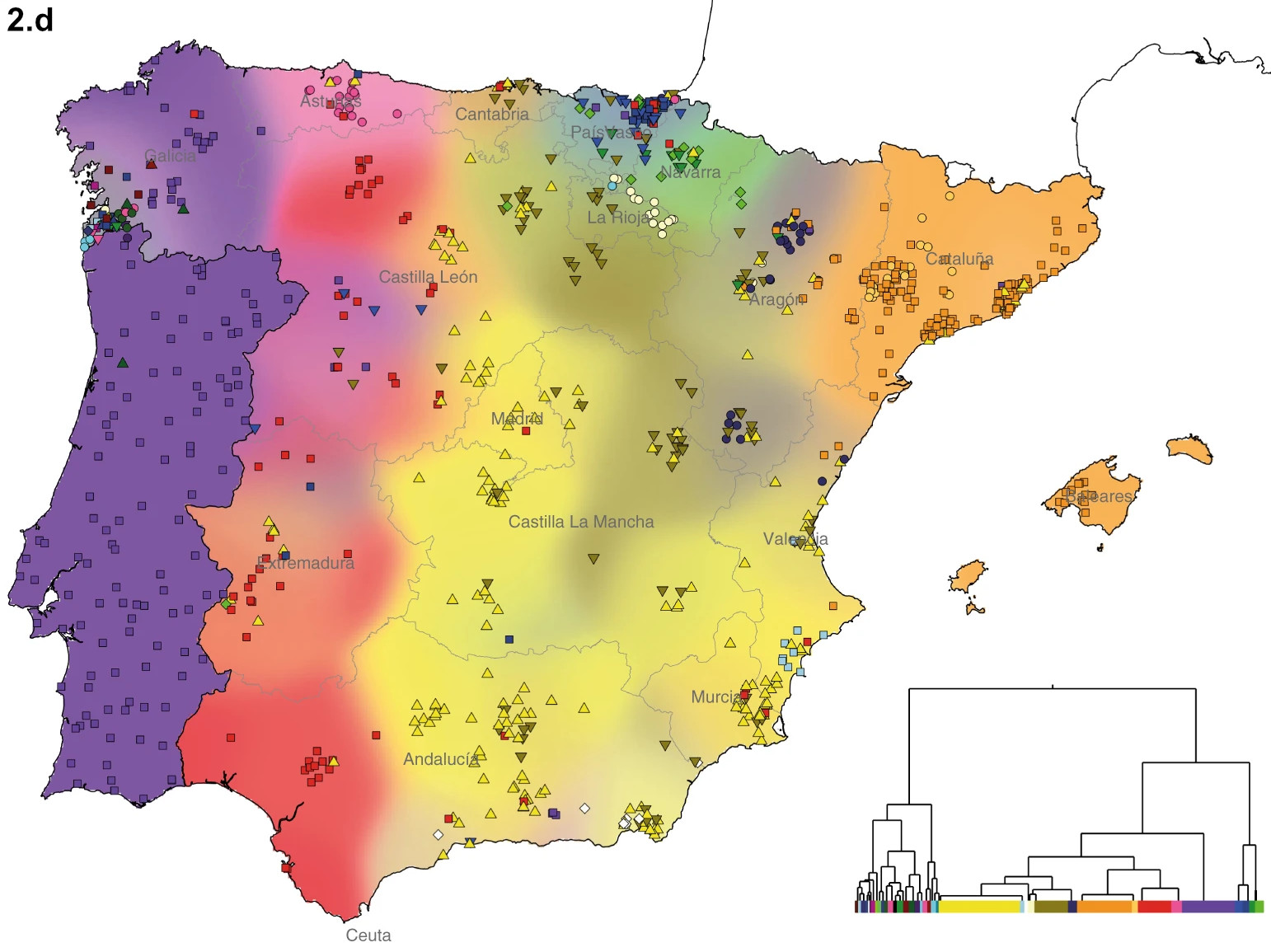
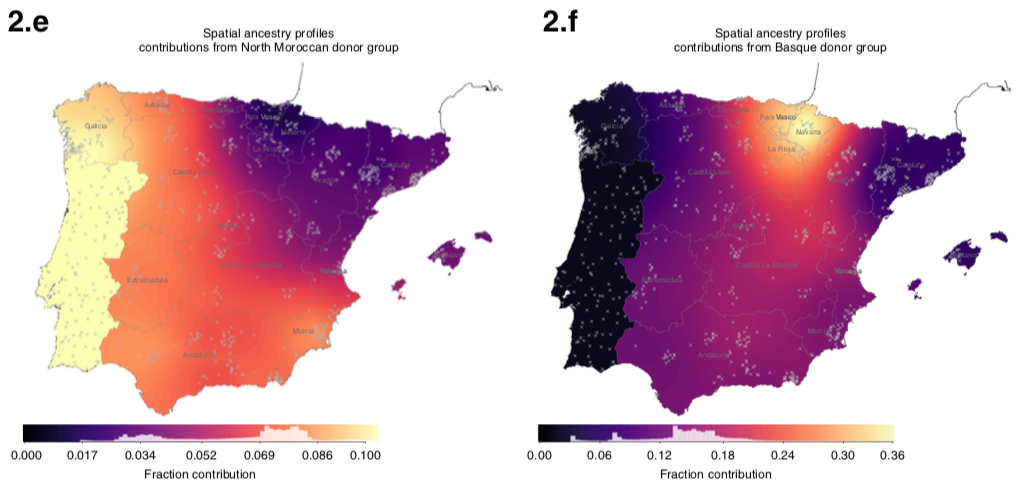
L'article entier est touffu mais passionnant, avec un élargissement sur l'influence des pays et régions limitrophes: au-delà des brassages entre les groupes ibériques définis plus tôt, les populations française, italienne, marocaine, irlandaise et de l'Ouest du Sahara sont celles qui contribuent le plus au patrimoine génétiques des habitants actuels de l'Espagne et du Portugal. Le Pays basque est aussi un cas intéressant: le profil génétique des groupes qui y sont associés montrent une implantation plus tardive dans la péninsule, à partir du XIIème siècle. On y perçoit une influence moindre des populations d'Afrique du Nord, et les groupes se sont beaucoup moins étendus vers le sud.
Les deux cartes suivantes illustrent ainsi la diffusion des profils génétiques ancestraux du Maroc (2.e) et du Pays basque(2.f) (plus les couleurs sont claires, plus la contribution est importante):
Pour prolonger la lecture, les articles originaux et leurs pièces supplémentaires:
- Genes mirror geography within Europe, Supplementary Materials
- Patterns of genetic differentiation and the footprints of historical migrations in the Iberian Peninsula, Supplementary Information
-
pour single nucleotide polymorphism. Watson et Crick ont montré que les 4 bases azotées adénine (A), cytosine (C), guanine (G) et thymine (T) constituant l'ADN ne peuvent former que des paires A-T et C-G (en fonction du nombres de liaisons hydrogène que les bases peuvent établir entre elles). L'ADN est constitué de deux brins hélicoïdaux enroulés, un changement d'un seul nucléotide (soit, d'une base) sur un brin correspond bien à une variation d'une paire sur la molécule entière, le nucléotide apparié étant le complémentaire du premier. ↩
-
les individus dotés d'un héritage mixte (au moins 2 pays différents relevés pour les 4 grands-parents) étaient écartés de l'étude: il s'agissait pour l'équipe de définir un niveau de "stabilité géographique", sur au moins deux générations. ↩
-
en anglais, principal component analysis. Pour être un peu plus précis (ou jargonneux, c'est selon): la PCA vise à décorréler les variables, autrement dit à les rendre orthogonales. Elle met en jeu les notions d'éléments propres et les fameuses matrices de covariance, qui interviennent quasi-systématiquement dans les comparaisons d'échantillons 2 à 2 comme étape préliminaires, quelque soit les méthodes statistiques déployées ensuite. Le peu traduisible coancestry matrix désigne couramment ces matrices dans les contextes de généalogie. ↩
-
le Romanche n'avait apparemment pas de part significative dans les données recueillies. ↩
-
encore faut-il que les données et méthodes mises à disposition pour l'évaluation par les pairs soient effectivement passées au crible. Des rétractations sont évidemment possibles une fois un article publié, si des points cruciaux ont échappé au comité de lecture. Le joli foutoir occasionné par l'article bidonné de Surgisphere sur le traitement du Covid-19 par un fameux anti-paludique est un bon exemple d'auteurs en mal de citation relus à la légère. ↩
-
comme évoqué plus tôt, la première étape passe là aussi par la construction d'une matrice de covariance, où chaque ligne décrit un échantillon en fonction de tous les autres. Les auteurs ont peint les chromosomes, d'après le nom du logiciel employé. Une référence, développée en grande partie à Bristol! ↩
-
d'où la couleur unie du pays sur cette seconde carte colorée, les points des individus concernés ayant été distribués aléatoirement sur la carte. L'échantillon pour cette seconde analyse portait sur 1530 individus au total. ↩